This article is intended as an overview of some unexpected or non-intuitive features of gases and flows, in the context of vacuum systems and special gaseous environments such as oxygen-free atmospheres. It is based on my experience with vacuum systems for scanning electron and scanning probe microscopy, as well as noble gas filled glovebox and sintering chamber.
An important concept in understanding gases is partial pressure. The key is that different gas species do not interact, so the presence or absence of one species makes no difference to the behavior of the other species. As a case study, consider tennis balls filled with argon, meant to last for a long time since argon does not diffuse out as readily. Over time they were seen to bulge and explode as the pressure inside them increased. Where did the extra pressure come from? Surely, it takes energy to pump gas up a pressure gradient, and what supplied this energy? The argon was pure, that is, absent of oxygen and nitrogen. Therefore even though the total pressure of argon inside the ball was higher than atmospheric, there was nonetheless a negative 1 atmosphere of partial pressure for oxygen and nitrogen, pushing them to enter the ball. The presence or absence of argon inside the ball does not have an impact on this partial pressure driving force. The oxygen and nitrogen then diffuse into the ball from the outside, raising the total pressure inside the ball beyond its intended design, leading it to explode. The extra energy to increase the internal pressure came from the work inherent in removing the partial pressure of oxygen and nitrogen from the volume inside the ball.
Another case study concerns boiling water. At room temperature, water has a partial pressure (vapor pressure) of about 0.02 atm, that is, 2% of total atmospheric pressure. If the partial pressure of water in air is lower than this, then liquid water is driven to evaporate at a certain rate which is defined by its temperature. This can be seen by placing a cup of water on a counter, and after a while noting that the cup becomes empty - the water has evaporated into the air and moved elsewhere. If the air is completely dry, that is it has a water partial pressure of 0.00 atm, it will remove all evaporating water, and still the water will not instantly evaporate or boil, but rather evaporate gradually, slower at low temperatures and faster at high temperatures. Yet it is a well known fact that at lower pressures the boiling point of water is lowered, and in a vacuum chamber it is possible to boil water even at room temperature. What is the difference between water in a vacuum chamber and water in very dry air? In both cases the partial pressure of water outside the liquid volume is zero, so the evaporation rate is equal, however in air the nitrogen and oxygen exert a force on the water which prevents bubbles from forming inside the liquid. In vacuum, there is no such force, so bubbles can form. Note that in a vacuum, bubbles have very little material inside them - they are visually impressive, but in terms of mass balance they are practically non-contributors, because it doesn't take much gas to form a big bubble in a vacuum (this must be kept in mind for vacuum casting applications). So even at an identical evaporation rate (per liquid surface area), in air the evaporation will not look impressive, while in a vacuum chamber it will be readily visible as bubbles form and break. (Clouds, similarly, are more visual than mass based phenomena, which is how they may seem to appear and disappear in the sky - the mass of water vapor in the air does not disappear but rather becomes transparent or opaque.) Temperature is what drives evaporation, so even vacuum dryers must heat the sample that is to be dried, the main advantage of the vacuum being that the water partial pressure can be made very low so the sample can be made very dry. Clothes dryers heat up the air that is blown over the clothes to speed up evaporation over what would happen on a clothes line.
When a sample is introduced into a high purity environment, such as the vacuum inside of a SEM, or the oxygen-free atmosphere in a sealed glovebox, some volume of the device (the air lock) will be exposed to the external atmosphere. Subsequently, the air from this volume needs to be removed so contaminants from the air are not transferred into the high purity environment. The two aforementioned examples may seem different because the SEM is at high vacuum while the glovebox is at atmospheric pressure (of, say, argon); however the process of removing the air is quite alike, because it is the partial pressure of the undesired species (primarily oxygen, water, nitrogen) that must be reduced - whether or not a fill of argon is then introduced into the air lock does not make a difference to the removal efficacy. For a high vacuum system, we connect the air lock to a vacuum pump and wait until the pressure is low enough that the undesired species are sufficiently sparse. For a glovebox, we may do the same and then fill the air lock with pure argon to reach atmospheric pressure, or we may remain at atmospheric pressure the whole time (avoiding the need for an expensive vacuum chamber) and continuously flow pure argon into the chamber while allowing the impure gas from the chamber to exit to the exhaust. Over time, given that the incoming argon is sufficiently pure, the volume of the chamber will tend towards the same purity, even though we never "pull" the undesired species out. Indeed a point of emphasis here is that it is not possible to "pull" gas molecules out as one cannot pull on a vacuum.
We may compare the above two approaches in their efficacy of removing contaminants as a function of pure gas volume used. For a continuous slow flow replacing the volume, the expression for remaining initial species is simply C=e-v, where v is the volume of freshly flowed gas (in units of the volume of the chamber). So, to reach C of 10%, v=2.3, that is, to replace 90% of the initially present gas with the pure gas, will require continuously flowing in 2.3x the chamber volume of the pure gas. Whereas if we had a vacuum pump that pumps down the chamber to 10% of atmospheric pressure, we could apply this vacuum pump and re-fill the chamber with the pure gas, then using only v=0.9 to reach the same result. Then if we use the same pump and re-fill again, we can reach C of 1% with v=1.8, repeating more as needed for higher purity. The graphic below shows a comparison with a few different vacuum pump effectiveness levels, where the blue and purple lines are representative of the previous sentences.
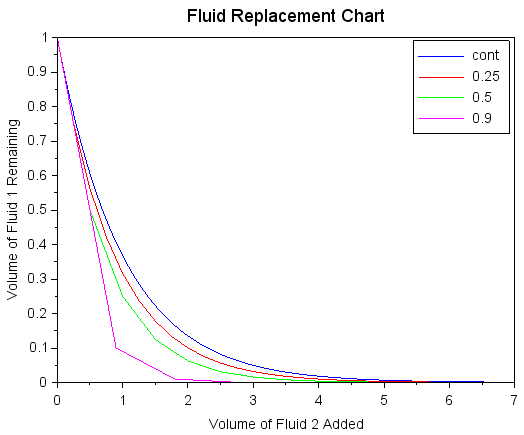
Comparison of the volumetric efficacy of replacing an initial volume of Fluid 1 with Fluid 2. The blue line represents a continuous flow of Fluid 2 into the chamber, while the other lines represent periodic removal of some volume fraction of the chamber (vacuum pump or dumping out liquid) and refill with Fluid 2.
As the purity requirements become more stringent, that is a lower C, the periodic removal approach becomes increasingly economic for minimizing the use of Fluid 2. Another aspect of periodic removal is that it is generally less susceptible to upstream contamination than a continuous flow, because in the periodic case only one valve to the chamber will be open at a time. There is nonetheless a caveat: the replacement ratios shown above are no higher than 0.9 while common oil-based rotary vacuum pumps can easily reach 0.999, and then it may seem that pumping down a few times with a vacuum pump will get arbitrarily low C, such as 0.001 with 1 replacement, 0.000001 with 2, 0.000000001 with 3, and so on. However the true limit on environment purity comes from the partial pressure of the contaminating species due to the pump, and diluting it with some pure gas does not change this at all. So if the pump connected to the chamber vents to the atmosphere, which is filled with oxygen, then even if we pump down (to the pump limit) and re-fill 10 times, the partial pressure of oxygen inside the chamber will be no better than the partial pressure which would have been there if we just pump down once (to the pump limit). The notion of diluting the contaminants is a decent rule of thumb on the macro scale, but at the microscopic level we cannot exceed the intrinsic limits of the pump by adding in any other gases, because once again it is not possible to "pull" gases from a volume or to "pull" on a vacuum.
Continuing the discussion of partial pressure, it must be recognized that the ideal gas model equivalently states that each gas molecule may be treated as completely non-interacting with other molecules but only with the apparatus walls. Thus the presence or absence of other gases in a volume makes no difference to how rapidly or how effectively some particular undesired gas will be removed. The only factor that makes a difference is the effectiveness of the vacuum pump in pulling all gases of that sort out of the volume and preventing them from coming back in, that is, the ultimate partial pressure that the pump can achieve, a function of its mechanical design. I still regularly see operating procedures for vacuum systems that suggest flowing argon (or some other pure gas) while pumping down an impure gas volume, with the intention of "flushing out" the undesirable gas with the argon flow, and I hope it is seen by now that this only wastes argon and does not at all help move the undesirable gas out faster. In fact, opening the argon valve while the chamber still contains impure gas exposes the system to a possibility of upstream contamination as the undesirable gas partial pressure in the chamber will be much higher than its partial pressure in the pure argon piping (even if the total pressure of argon is higher - but again this doesn't matter because gases do not interact), so this will present a driving force for the undesirable gas to enter the pure argon piping and diffuse further upstream basically uninhibited by the flow of argon in the opposite direction (molecules are moving very fast just from their thermal energy, so a little bit of a flow in a pipe will not prevent upstream diffusion especially in the molecular regime). The diffused impure gas can then leak back into the chamber over a long time, upsetting the experimental conditions and being difficult to trace. The proper approach is to connect the vacuum pump to the chamber and let it pump down to the pump's limit, then open the argon valve with the purpose of flushing out possible contaminants in the argon line assuming it has at some point been exposed to the atmosphere again reaching the pump's limit, and only then open the argon tank so there is minimal chance of contaminants entering the tank volume. Any deficiencies should be remediated by using a better vacuum pump rather than by "flushing" or "dilution".
Still for reaching high vacuum levels, as inside a SEM, the limit is generally not the ultimate partial pressure of the vacuum pump, but the very slow release of undesired molecules adhered to chamber surfaces. While flowing an inert gas will make no difference to this rate as described above (except, notably, it may appear to improve the vacuum in gas-sensitive vacuum gauges, where an argon background may give an illusory appearance of better vacuum than a nitrogen background), heating the chamber surfaces will increase the rate. (Flowing an appropriate chemically reactive gas may capture the undesired molecules and increase this rate, but I have rarely seen this used) This is the rationale for baking out high vacuum systems, raising surface temperatures to above 200 C, so that adsorbed molecules will release and be pumped by the vacuum pump, and the ultimate vacuum may be reached after a few days rather than a few months. Water is a major contaminant, and the water molecule has a particularly high affinity for clean metal surfaces, so I expect that appropriately functionalized chamber surfaces (such that they are hydrophobic) will make a big impact on the pump down time, however I have not seen this attempted at scale.
With a flowing stream of gas or liquid, our intuition is that everything will move in the direction of the flow, like an "ideal pump", however at purity levels having contaminants measured in ppm and lower, the ability of contaminants to flow upstream against the macroscopic flow must not be ignored. With gases and vacuum systems, the ability of molecules to travel a long distance up a substantial macroscopic pressure gradient must not be overlooked (physically small orifices which are gas "chokepoints" (based on local speed of sound) act as the main barrier, while large diameter pipes are no impediment even if long). A water bubbler is a device that breaks up a macroscopic flow into individual bubbles imposing some required directionality to the flow of contaminants, however this will be limited by the solubility of the contaminant in water and its ability to diffuse along surfaces, including along the surfaces of the bubbler (even those immersed in water) towards the clean side of the system. Even with something as simple as pouring water from one beaker to another, contaminants may flow upwards into the beaker out of which water is being poured, due to surface tension or by small droplets formed at the water surface. Liquids having lower solubility of the contaminants, chemically reactive species that capture the contaminants, and cold surfaces that condense the contaminants, are some options for avoiding upstream contamination.